Using BiVariAn solves several different problems, each one described in its corresponding function.
Functions
There are currently several groups of functions available:
Database oriented
encode_factorsfix_mixed_cols
Plotting oriented
theme_serenetheme_serene_voidauto_bar_categauto_bar_contauto_viol_contauto_bp_contauto_corr_contauto_dens_contauto_pie_categ
Univariate and bivariate analyses
auto_shapiro_rawdichotomous_2k_2sidcontinuous_2gcontinuous_2g_paircontinuous_corr_testcontinuous_multgcontinuous_multg_rm
Regression analyses
step_bw_pstep_bw_firthlogistf_summaryss_multreg
Usage examples
Suppose we want to start an analysis of a dataset with a large number of variables. Typically, we would have to run normality tests, which would mean writing one line of code for every variable. As an example, we will use the penguins dataset available in the palmerpenguins package.
data(penguins)
dat <- penguinsNormality
If we inspect the dataset we have 8 variables with 344 observations. Among these variables there are 5 numeric variables (one of them representing years), which would mean writing normality test code for each of them.
shapiro.test(dat$bill_len)
#>
#> Shapiro-Wilk normality test
#>
#> data: dat$bill_len
#> W = 0.97485, p-value = 1.12e-05More advanced users might consider writing a loop or using helper functions such as lapply to evaluate all variables through a list (or similar approaches).
cont_var <- c("bill_len", "bill_dep", "flipper_len", "body_mass")
lapply(dat[cont_var], function(x) shapiro.test(x))
#> $bill_len
#>
#> Shapiro-Wilk normality test
#>
#> data: x
#> W = 0.97485, p-value = 1.12e-05
#>
#>
#> $bill_dep
#>
#> Shapiro-Wilk normality test
#>
#> data: x
#> W = 0.97258, p-value = 4.419e-06
#>
#>
#> $flipper_len
#>
#> Shapiro-Wilk normality test
#>
#> data: x
#> W = 0.95155, p-value = 3.54e-09
#>
#>
#> $body_mass
#>
#> Shapiro-Wilk normality test
#>
#> data: x
#> W = 0.95921, p-value = 3.679e-08With BiVariAn we can solve this problem (and improve the display of the results) by using the auto_shapiro_raw function.
auto_shapiro_raw(data = dat)Variable |
p_shapiro |
Normality |
|---|---|---|
bill_len |
<0.001* |
Non-normal |
bill_dep |
<0.001* |
Non-normal |
flipper_len |
<0.001* |
Non-normal |
body_mass |
<0.001* |
Non-normal |
year |
<0.001* |
Non-normal |
Do we want a data frame instead of a flextable? We can do that with the flextableformat argument (which is used in most functions).
shapirores <- auto_shapiro_raw(
data = dat,
flextableformat = FALSE
)
shapirores
#> Variable p_shapiro Normality
#> bill_len bill_len <0.001* Non-normal
#> bill_dep bill_dep <0.001* Non-normal
#> flipper_len flipper_len <0.001* Non-normal
#> body_mass body_mass <0.001* Non-normal
#> year year <0.001* Non-normalBivariate analyses
After checking normality, the next natural step is to apply bivariate tests, but again we would have to write separate code for each test. For example, if we wanted to compare variables across sexes, we would write something like this for every variable.
Note
The normality analysis showed a non normal distribution, so ideally at this stage we would not apply parametric tests. For now, we will continue as if we needed to perform a t test, only to illustrate the workflow.
# First we would evaluate homoscedasticity
car::leveneTest(
dat$bill_len,
group = dat$sex,
center = "median"
)
#> Levene's Test for Homogeneity of Variance (center = "median")
#> Df F value Pr(>F)
#> group 1 2.8244 0.09378 .
#> 331
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
# Then we apply the t test
t.test(dat$bill_len)
#>
#> One Sample t-test
#>
#> data: dat$bill_len
#> t = 148.78, df = 341, p-value < 2.2e-16
#> alternative hypothesis: true mean is not equal to 0
#> 95 percent confidence interval:
#> 43.34125 44.50261
#> sample estimates:
#> mean of x
#> 43.92193Doing this for every variable quickly becomes an iterative and repetitive process, and in addition we would need to write separate code to report mean differences with their corresponding tests.
With BiVariAn we can avoid this by using the continuous_2g function.
continuous_2g(
data = dat,
groupvar = "sex"
)Variable |
P_Shapiro_Resid |
P_Levene |
P_T_Test |
Var_Equal |
P_Mann_Whitney |
Diff_Means |
CI_Lower |
CI_Upper |
Significant_test |
|---|---|---|---|---|---|---|---|---|---|
bill_len |
<0.001* |
0.09 |
<0.001* |
true |
<0.001* |
-3.76 |
-4.87 |
-2.65 |
Mann-W-U test |
bill_dep |
<0.001* |
0.69 |
<0.001* |
true |
<0.001* |
-1.47 |
-1.86 |
-1.07 |
Mann-W-U test |
flipper_len |
<0.001* |
0.04 |
<0.001* |
false |
<0.001* |
-7.14 |
-10.06 |
-4.22 |
Mann-W-U test |
body_mass |
<0.001* |
0.01 |
<0.001* |
false |
<0.001* |
-683.41 |
-840.58 |
-526.25 |
Mann-W-U test |
year |
<0.001* |
1.00 |
0.99323 |
true |
0.99373 |
0.00 |
-0.17 |
0.18 |
None |
Customizing the output
Similarly, we can use the flextableformat argument to obtain a data frame.
continuous_2g(
data = dat,
groupvar = "sex",
flextableformat = FALSE
)
#> Variable P_Shapiro_Resid P_Levene P_T_Test Var_Equal P_Mann_Whitney
#> 1 bill_len <0.001* 0.09378 <0.001* TRUE <0.001*
#> 2 bill_dep <0.001* 0.68955 <0.001* TRUE <0.001*
#> 3 flipper_len <0.001* 0.04011 <0.001* FALSE <0.001*
#> 4 body_mass <0.001* 0.01435 <0.001* FALSE <0.001*
#> 5 year <0.001* 0.99834 0.99323 TRUE 0.99373
#> Diff_Means CI_Lower CI_Upper Significant_test
#> 1 -3.75779 -4.86656 -2.64903 Mann-W-U test
#> 2 -1.46562 -1.86021 -1.07103 Mann-W-U test
#> 3 -7.14232 -10.06481 -4.21982 Mann-W-U test
#> 4 -683.41180 -840.57826 -526.24533 Mann-W-U test
#> 5 0.00076 -0.17478 0.17630 NoneHowever, we can also add labels to the variables using the table1::label() function from the table1 package.
And we obtain a nicely labeled table.
continuous_2g(
data = dat,
groupvar = "sex"
)Variable |
P_Shapiro_Resid |
P_Levene |
P_T_Test |
Var_Equal |
P_Mann_Whitney |
Diff_Means |
CI_Lower |
CI_Upper |
Significant_test |
|---|---|---|---|---|---|---|---|---|---|
Bill length |
<0.001* |
0.09 |
<0.001* |
true |
<0.001* |
-3.76 |
-4.87 |
-2.65 |
Mann-W-U test |
Bill depth |
<0.001* |
0.69 |
<0.001* |
true |
<0.001* |
-1.47 |
-1.86 |
-1.07 |
Mann-W-U test |
flipper_len |
<0.001* |
0.04 |
<0.001* |
false |
<0.001* |
-7.14 |
-10.06 |
-4.22 |
Mann-W-U test |
body_mass |
<0.001* |
0.01 |
<0.001* |
false |
<0.001* |
-683.41 |
-840.58 |
-526.25 |
Mann-W-U test |
year |
<0.001* |
1.00 |
0.99323 |
true |
0.99373 |
0.00 |
-0.17 |
0.18 |
None |
Do we want a title row that indicates the grouping variable? We can add it with the caption argument.
continuous_2g(
data = dat,
groupvar = "sex",
caption = TRUE
)Sex | |||||||||
|---|---|---|---|---|---|---|---|---|---|
Variable |
P_Shapiro_Resid |
P_Levene |
P_T_Test |
Var_Equal |
P_Mann_Whitney |
Diff_Means |
CI_Lower |
CI_Upper |
Significant_test |
Bill length |
<0.001* |
0.09 |
<0.001* |
true |
<0.001* |
-3.76 |
-4.87 |
-2.65 |
Mann-W-U test |
Bill depth |
<0.001* |
0.69 |
<0.001* |
true |
<0.001* |
-1.47 |
-1.86 |
-1.07 |
Mann-W-U test |
flipper_len |
<0.001* |
0.04 |
<0.001* |
false |
<0.001* |
-7.14 |
-10.06 |
-4.22 |
Mann-W-U test |
body_mass |
<0.001* |
0.01 |
<0.001* |
false |
<0.001* |
-683.41 |
-840.58 |
-526.25 |
Mann-W-U test |
year |
<0.001* |
1.00 |
0.99323 |
true |
0.99373 |
0.00 |
-0.17 |
0.18 |
None |
Important
To use the
captionargument it is mandatory thatflextableformatis TRUE; otherwise an error will be raised.
Graphical representation
The next step would be to create graphical representations. Again, if we want to use ggplot2 we would write separate code for every plot (which implies multiple lines of code that can become tedious if we want extensive customization).
Let us create a boxplot to represent the comparison of flipper_len across the sex variable.
dat_comp <- dat %>%
drop_na(sex) # We remove the NAs from the sex variable
ggplot(
data = dat_comp,
mapping = aes(
x = sex,
y = flipper_len
)
) +
geom_boxplot() +
labs(
x = "Sex",
y = "Flipper length"
) +
theme_classic()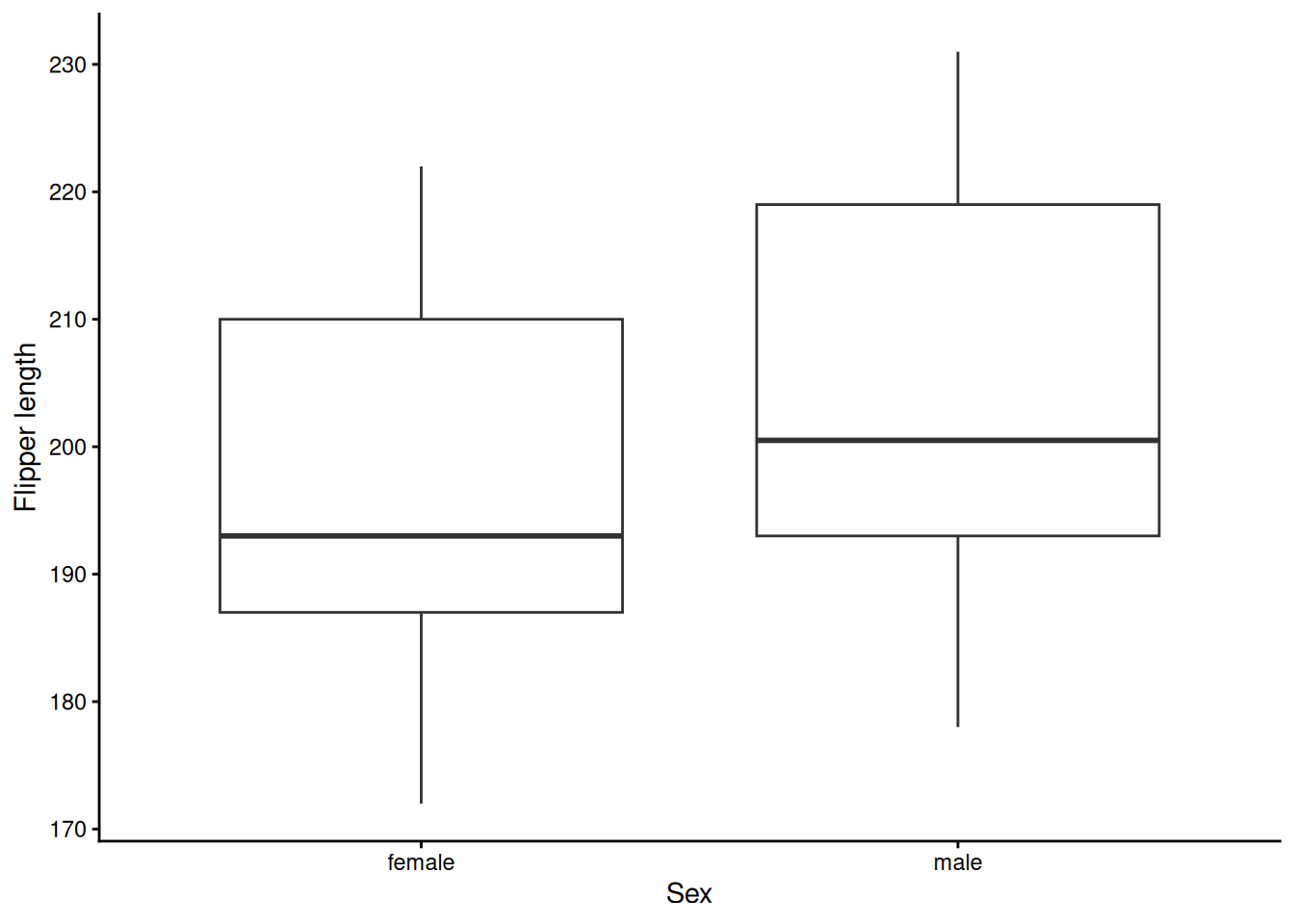
Repeating this for every variable implies multiple lines of code. For this reason, the auto_bp_cont function can help streamline the workflow.
We simply call the function and store its output in an object. This object is a list, so we can select the specific plot we want to display or include in our report.
graphs <- auto_bp_cont(
data = dat_comp,
groupvar = "sex"
)
graphs$flipper_len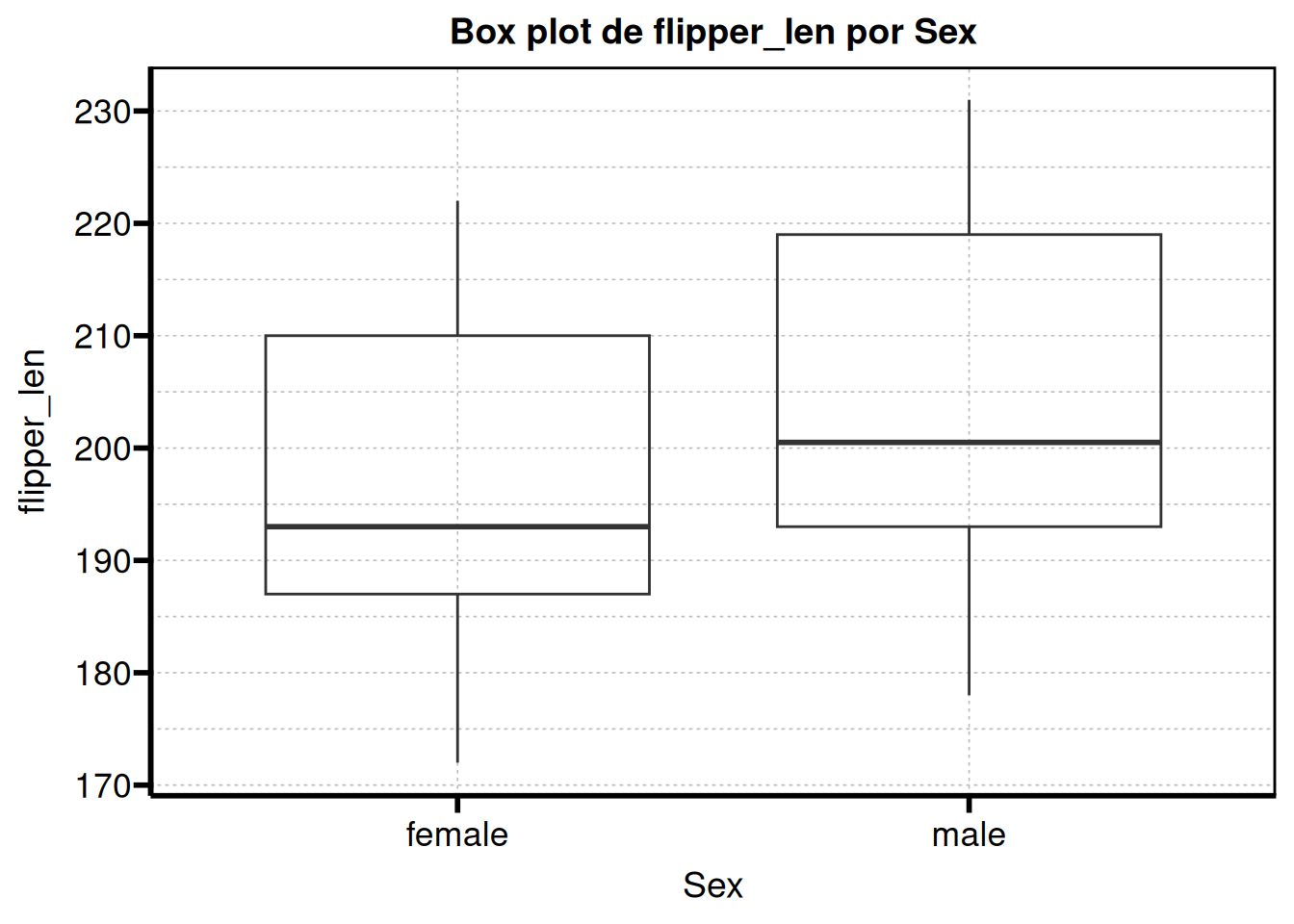
If you remember, we previously assigned labels using table1::label(). The good news is that these labels can also be used for the plots.
table1::label(dat_comp$sex) <- "Sex"
table1::label(dat_comp$flipper_len) <- "Flipper length"
graphs <- auto_bp_cont(
data = dat_comp,
groupvar = "sex"
)
graphs$flipper_len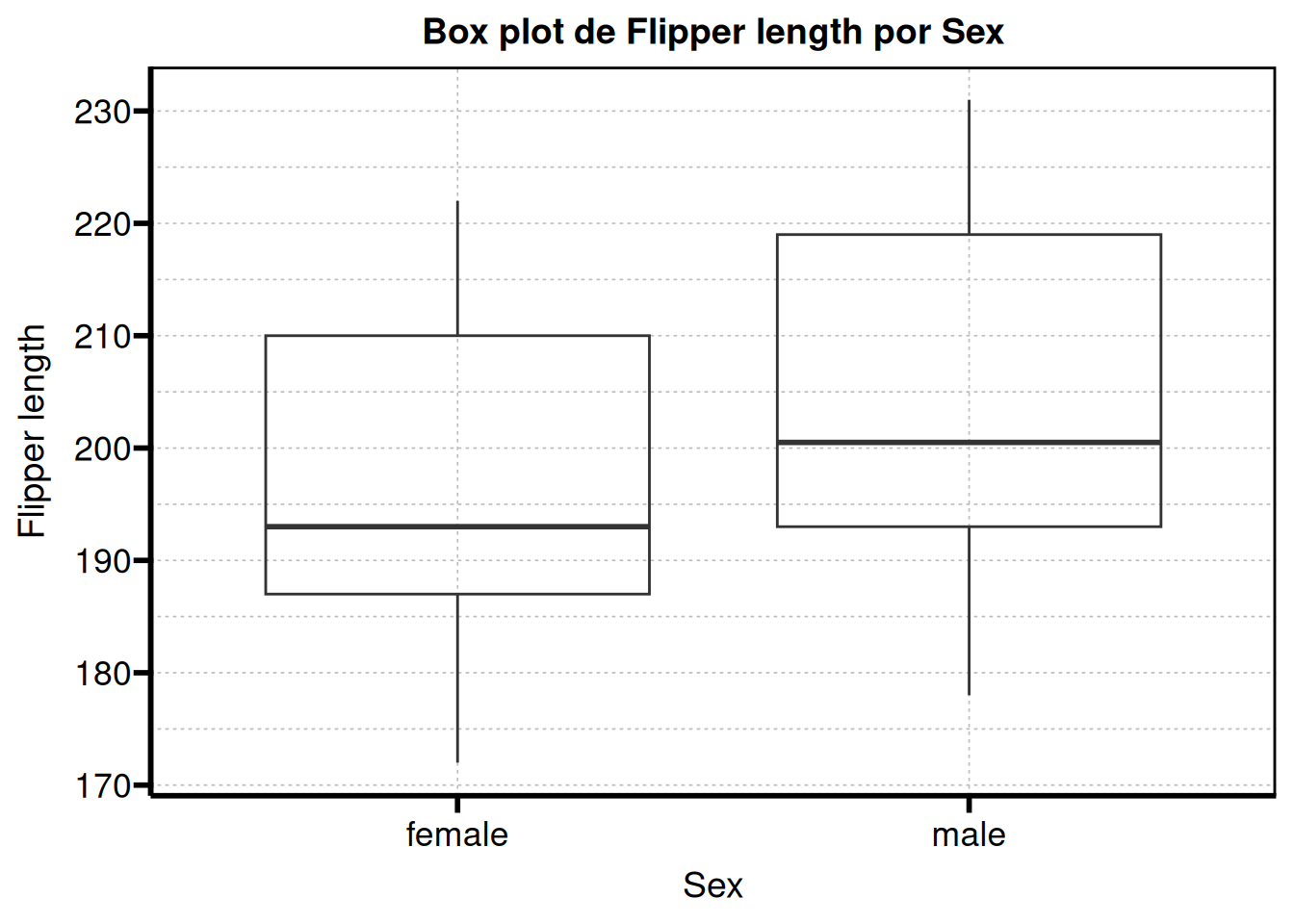
At this point we have everything we need to build our report.
Special mentions
As a special mention, there are some useful helper functions for regression analysis. For example, the step_bw_p function helps with the tedious iterative process of simplifying regression models by stepwise backward selection based on a p value threshold, and the step_bw_firth function performs the same process for models fitted with logistf::logistf().
If we fit a model with our dataset, we obtain something like this:
model_reg <- glm(
sex ~ species + island + bill_len + bill_dep + flipper_len + body_mass,
data = dat,
family = binomial()
)
step_bw_p(model_reg)
#>
#> Initial model: sex ~ species + island + bill_len + bill_dep + flipper_len + body_mass
#> Analysis of Deviance Table (Type II tests)
#>
#> Response: sex
#> LR Chisq Df Pr(>Chisq)
#> species 32.685 2 7.991e-08 ***
#> island 1.061 2 0.5882
#> bill_len 32.923 1 9.588e-09 ***
#> bill_dep 35.316 1 2.803e-09 ***
#> flipper_len 0.304 1 0.5815
#> body_mass 50.238 1 1.362e-12 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> Removing island (p = 0.5882)
#> Analysis of Deviance Table (Type II tests)
#>
#> Response: sex
#> LR Chisq Df Pr(>Chisq)
#> species 31.895 2 1.186e-07 ***
#> bill_len 33.097 1 8.769e-09 ***
#> bill_dep 34.748 1 3.752e-09 ***
#> flipper_len 0.189 1 0.6638
#> body_mass 51.667 1 6.575e-13 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> Removing flipper_len (p = 0.6638)
#> Analysis of Deviance Table (Type II tests)
#>
#> Response: sex
#> LR Chisq Df Pr(>Chisq)
#> species 32.590 2 8.378e-08 ***
#> bill_len 33.727 1 6.343e-09 ***
#> bill_dep 36.486 1 1.538e-09 ***
#> body_mass 58.852 1 1.700e-14 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> No eliminable terms above p_threshold. Stopping.
#> $final_model
#>
#> Call: glm(formula = sex ~ species + bill_len + bill_dep + body_mass,
#> family = binomial(), data = dat)
#>
#> Coefficients:
#> (Intercept) speciesChinstrap speciesGentoo bill_len
#> -76.093935 -6.897694 -7.875212 0.620082
#> bill_dep body_mass
#> 1.643613 0.005962
#>
#> Degrees of Freedom: 332 Total (i.e. Null); 327 Residual
#> (11 observations deleted due to missingness)
#> Null Deviance: 461.6
#> Residual Deviance: 127.3 AIC: 139.3
#>
#> $steps
#> Step
#> 1 Initial
#> 2 - island
#> 3 - flipper_len
#> Formula
#> 1 "sex ~ species + island + bill_len + bill_dep + flipper_len + body_mass"
#> 2 "sex ~ species + bill_len + bill_dep + flipper_len + body_mass"
#> 3 "sex ~ species + bill_len + bill_dep + body_mass"
#>
#> attr(,"class")
#> [1] "step_bw"In practice, this removes the need to manually perform three backward selection steps (which would otherwise require typing the full model each time or using helpers such as reformulate).
We can also store the output in an object:
step_res <- step_bw_p(
model_reg,
trace = FALSE # To avoid printing the process in the console
)
step_res$steps # Data frame of the steps involved
#> Step
#> 1 Initial
#> 2 - island
#> 3 - flipper_len
#> Formula
#> 1 "sex ~ species + island + bill_len + bill_dep + flipper_len + body_mass"
#> 2 "sex ~ species + bill_len + bill_dep + flipper_len + body_mass"
#> 3 "sex ~ species + bill_len + bill_dep + body_mass"
step_res$final_model # Final model after all iterations
#>
#> Call: glm(formula = sex ~ species + bill_len + bill_dep + body_mass,
#> family = binomial(), data = dat)
#>
#> Coefficients:
#> (Intercept) speciesChinstrap speciesGentoo bill_len
#> -76.093935 -6.897694 -7.875212 0.620082
#> bill_dep body_mass
#> 1.643613 0.005962
#>
#> Degrees of Freedom: 332 Total (i.e. Null); 327 Residual
#> (11 observations deleted due to missingness)
#> Null Deviance: 461.6
#> Residual Deviance: 127.3 AIC: 139.3